Quantum espresso 같은 DFT 방법의 주요 단점 중 하나는 밴드 갭을 과소 평가한다는 것이다. 이러한 과소평가는 자기 상호작용 에너지의 오류 때문에 발생하는 것으로 알려져 있다.
이러한 오류로 전자가 지나치게 비편재화되어 있는 것으로 계산되어 점유된 스테이트의 애너지가 위쪽으로 말려나게되고, 반도체 및 절연체의 밴드갭이 실제보다 훨씬 낮게 계산되는 결과를 가져온다.
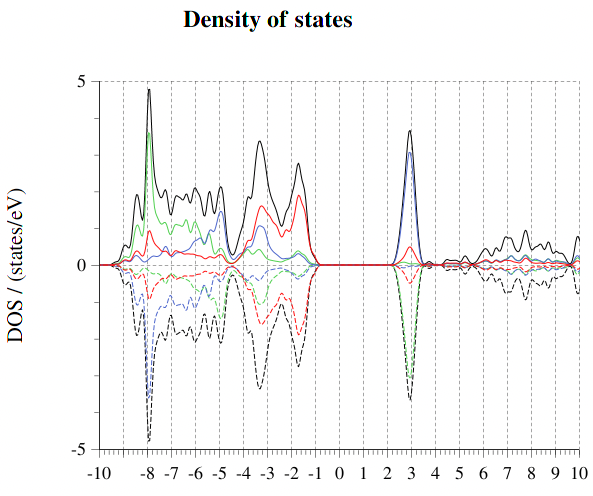
예를 들자면 quantum Espresso로 계산한 NiO의 DOS응 다음과 같은데 밴드갭이 약 0.8 고 계산된다.
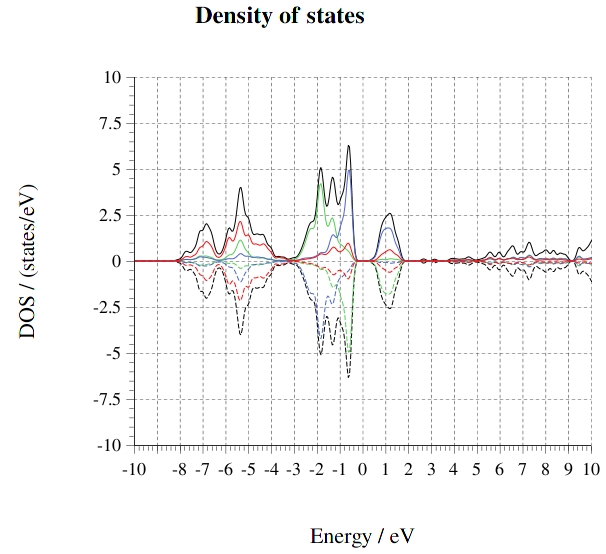
이러한 문제를 해결하는 한방법으로 Hubbard 보정법이 있다. 이 방법 (DFT+U) 에서는 점유 스테이트가 지역화하도록 추가로 준실험적으로 설정된 포텐셜에너지(U)를
부가하여 주는데, d- 또는 f- orbital의 전자를 갖는 원소에 적용할 때 특히 좋은 결과를 보여 주는 것으로 알려져 있다.
LDA+U 계산을 하려면 "system" 에 다음을 포함시켜야 한다.
&system
.
.
lda_plus_u = .true.,
lda_plus_u_kind = $Utype,
Hubbard_U(1) = $U1.
Hubbard_U(2) = $U2,
.
.
/여기서 $Utype 에는 0,1, 그리고 2가 가능한데
lda_plus_u_kind=0 : Cococcioni and de Gironcoli, PRB 71, 035105 (2005) 가 제시한 방법을 단수화 시킨 방법으로, Hubbard_U 변수 값을 설정하여 사용한다.
lda_plus_u_kind=1 : Liechtenstein et al., 등이 제시한 방법으로 Hubbard_U and Hubbard_J 의 값을 설정하여 사용한다.
lda_plus_u_kind=2 : Hubbard_U and Hubbard_V 의 값을 설정하여 사용한다.
원자에 따라 $U1, $U2 값을 다르게 적용하는데 $U1, $U2 는 eV 단위의 숫자로 일반적으로 7-8 eV 이하의 값을 갖고 보통 0 < $U1, $U2 < 5 eV 이다.
NiO 를 예로 들자면
lda_plus_u=.true.,
lda_plus_u_kind = 0,
Hubbard_U(1)=7.6,
Hubbard_U(2)=7.6,을 &system 에 포함시켜 준다.
NiO lda+U 계산을 위한 full input file
&control
calculation='scf' ,
prefix='NiO' ,
restart_mode='from_scratch' ,
tstress = .false.
tprnfor = .true.
wf_collect=.false. ,
pseudo_dir='/Users/Work/QE/PPots/' ,
outdir='/Users/Work/QE-test/NiO/NiO-u7.6/scratch/' ,
verbosity = 'high'
/
&system
ibrav = 5 ,
celldm(1) =9.67155,
celldm(4)=0.8333333333,
nat = 4 ,
ntyp = 3 ,
nspin=2,
occupations = "tetrahedra"
ecutwfc =50
starting_magnetization(1)=1.0,
starting_magnetization(2)=-1.0,
tot_magnetization = 0.0
nbnd=52,
lda_plus_u=.true.,
lda_plus_u_kind = 0,
Hubbard_U(1)=7.6,
Hubbard_U(2)=7.6,
/
&electrons
diagonalization = 'david'
mixing_mode = 'plain'
mixing_beta = 0.3
conv_thr = 1.e-8
/
ATOMIC_SPECIES
Ni 58.69340 ni_pbesol_v1.4.uspp.F.UPF
NiB 58.69340 ni_pbesol_v1.4.uspp.F.UPF
O 15.99940 O.pbesol-n-kjpaw_psl.0.1.UPF
ATOMIC_POSITIONS crystal
Ni 0.0000000000 0.0000000000 0.0000000000
NiB 0.5000000000 0.5000000000 0.5000000000
O 0.2500000000 0.2500000000 0.2500000000
O 0.7500000000 0.7500000000 0.7500000000
K_POINTS automatic
6 6 6 0 0 0